Embedded Files
IMPORTANT: This is the legacy GATK documentation. This information is only valid until Dec 31st 2019. For latest documentation and forum click here
IMPORTANT: This is the legacy GATK documentation. This information is only valid until Dec 31st 2019. For latest documentation and forum click here
created by samwell
on 2017-12-21
By Sam Friedman, deep learning developer in GATK4
Over the past couple of weeks, there's been a lot of chatter online --and in the press!-- about the applicability of deep learning to variant calling. This is exciting to me because I've been working on developing a deep learning approach to variant filtering based on Convolutional Neural Networks (CNNs), with the goal of replacing the VQSR filtering step in the GATK germline short variants pipeline. In fact, multiple groups have been picking up on the promise of deep learning and applying it to genomics.
As far as I'm aware, the first group to publish a deep learning-based approach for variant calling was the Campagne lab at Cornell, who released their variationanalysis software in December 2016. There's also a group at Illumina that has been doing some interesting work with deep learning for predicting functional effects of variants, and some of my colleagues are currently working with researchers at Microsoft to see if CNNs can be used to discover complex Structural Variations (SVs) from short sequencing reads.
When the Google team made the source for DeepVariant public last week, I ran a few tests to compare it to the tool that I've been working on for the last six months (GATK4 CNN). The results are summarized in the table below.

Both my tool and DeepVariant outperform VQSR (our "traditional" variant filtering algorithm), especially when VQSR is run on a single sample rather than on a cohort according to our current best practices. The delta isn't all that large on SNPs, but that's expected because germline SNPs are largely a solved problem, so all tools tend to do great there. The harder problem is indel calling, and that's where we see more separation between the tools. The good news is we're all doing better than VQSR on calling indels, which means progress for the research community, whatever else happens. It doesn't hurt my mood that these preliminary findings suggest GATK4 CNN is doing even better than DeepVariant :)
But keep in mind it's early days yet for deep learning in genomics, so a lot could still change as we all figure out how best to take advantage of these methods. Read on if you want to know more about how these results were generated and how my tool works under the hood.
The table above was created from a held out test set of variants enriched for difficult calls. We compute the ROC curves from the VQSLOD scores from VQSR, QUAL scores from deep variant, and the scores from our CNN. SynDip, if you're not familiar with it, is the new truth set we're using as recently described by my colleague Yossi Farjoun here, and gnomAD is the Genome Aggregation Database produced by Daniel MacArthur and colleagues.
I'll be giving a talk about this work at the AGBT conference in Orlando in February 2018, and I'm aiming to have the tools ready for general release at that time. If you'd like to know more in the meantime, read on below, and feel free to check out the code on the GATK repo on Github.
How it works under the hood
How it works under the hood
Deep learning is a pretty rich field of research that comprises many different approaches, and there's still a lot to be figured out in terms of what's going to transfer well to any particular problem. For example, the Campagne lab's approach starts from reads and, in their own words, "maps structured data (alignment) to a small number of informative features, which can be used for efficient DL model training". In my work I've chosen to treat the problem as a variant filtering question (or classification in machine learning terms), where I take the output of the caller and use a deep learning process to filter that callset. It seems the Google team's approach is similar in that their software involves a first step that looks a lot like the HaplotypeCaller (which makes sense since Mark and Ryan wrote most of the HC's original code), then applies the deep learning classifier to the variant candidates.
My approach uses variants and annotations encoded as tensors, which carry the precise read and reference sequences, read flags, as well as base and mapping qualities. The modeling architecture I use is specifically designed to process these read tensors, and so avoids the extensive use of maxpooling found in many image classification CNNs (maxpooling is motivated by the smoothness prior of natural images, where neighboring pixels tend to be the same, but DNA sequences are not smooth, so we reserve maxpooling for the deeper layers of the net). Because the tensor dimensions contain different types of data, I convolve each separately so as to allow deep layers in the net to see this meaningful difference. This architecture also integrates annotations seamlessly into the classification, which accelerates learning times dramatically and opens the door to unsupervised training down the road.
The animations below shows two read tensors one containing a heterozygous SNP the other with a homozygous deletion of a short tandem repeat. You can see the reads with the individual base qualities, the reference sequence, the read flags and the mapping quality each in their own channel.


If you'd like to know more or if you're a developer yourself and want to discuss the pros and cons of various approaches, please feel free to comment in the discussion thread below.
Updated on 2019-04-10
From akundaje on 2017-12-22
What’s your class imbalance for your classification tasks. I expect there is significant imbalance since variants are rare events as compared to the size of the genome. If that is the case, auROC can be very misleading. You’d want to report area under the precision recall curve and recall at specific precisions eg. 70, 80, 90 %.
Had posted this on Twitter. You guys should get on there.
-Anshul Kundaje.
From samwell on 2017-12-22
Hi Anshul,
Thanks for the comment. The AUC table was made from a test set of variants enriched for negative examples so that the classes are nearly balanced.
-Sam
From akundaje on 2017-12-22
Ok great. But does your test set reflect the actual imbalance you expect to see when you use this to call variants across the whole genome? If not, then the test set would not be representative.
From samwell on 2017-12-23
Good point! We look only at sites emitted by HaplotypeCaller, not over the whole genome. Our variant filtering architecture classifies these calls as either real sites of variation or incorrect calls. We think AUC is still a valuable metric in this context, because HaplotypeCaller’s mandate is sensitivity and artifactual calls are not so rare. Of course, we also look at Precision and Recall Curves. Below you can see a table of area under the precision recall curve on the same test set as auROC above.
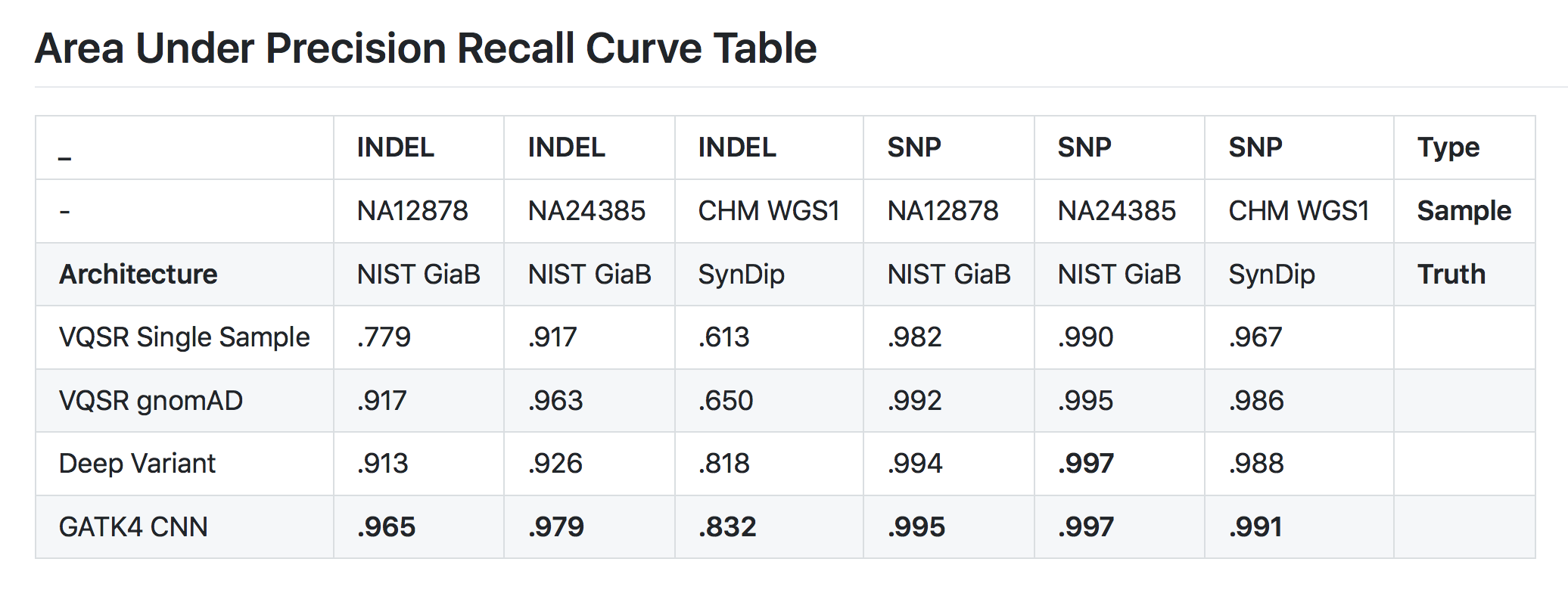
Class balancing is often tricky and it is especially difficult in genomics. In variant filtration, we have many fewer negative examples than positive ones and typically far fewer INDELs than SNPs. To avoid learning this background distribution, during training we uniformly sample from each of the classes in each minibatch. For variant calling, where the class imbalance is even more extreme, we have resorted to using a weighted categorical crossentropy as our loss function, which penalizes errors at sites of variation far more than errors at homozygous reference sites.
We are always looking for better ways to cope with the class balance problem, if you (or any reader) has ideas, please share!
From akundaje on 2017-12-23
Looks great. We use similar strategies with forcibly adding minority class samples to each minibatch and weighted loss. But still trying to figure out a loss function that explicitly optimizes precision/recall rather than auROC or cross entropy. Not there yet. Surprisingly few elegant direct solutions to the label imbalance problem.
From anuar12 on 2018-01-03
If one uses binary cross-entropy loss and optimizes for F1-score, you can try modifying binary cross-entropy to -log^2(x) or log^3(x), this way the loss would penalize wrong predictions after 0.5 more since F1 operates on discrete classes. And penalty for points that are closer to correct probability will be lower wrt original binary cross-entropy.
From ambrex on 2018-01-05
Nice animations of the tensors. Would you mind sharing what tool you used to create them?
From shlee on 2018-01-05
HI @ambrex,
I’ll let the developer know you have a question. It may be a while before they respond. We are gearing up for the GATK4 release next week.
From samwell on 2018-01-05
Hi @ambrex
Glad you liked the tensor animations! We wrote our own viewer. You can check out the [source code here](https://github.com/broadinstitute/dsde-deep-learning/tree/master/TensorViewer “source code here”). It is built in Java on top of the [processing](http://processing.org “processing”) library.
From Katie on 2018-01-24
This looks really interesting – will this be applicable for haploid /non-model organisms too? Thank you!
From Geraldine_VdAuwera on 2018-01-24
Hi Katie, we’re hopeful that this approach will eventually benefit a wide range of use cases down the road. See [this video snippet](https://youtu.be/ZZj--Y3oUQc?t=1h15m58s) from the GATK4 launch event where I asked samwell a question about this.
From Katie on 2018-01-24
Thanks, will check it out!
From daquang on 2018-01-31
Will you provide source code and technical details behind GATK CNN?
From Geraldine_VdAuwera on 2018-01-31
@daquang Of course! The source code is already publicly available in the GATK4 github repository. The developer in charge, Sam Friedman, will give a talk about it at AGBT in a couple of weeks, and we intend to document it further once the tools are out of beta phase.
From Katie on 2018-03-08
Hi – I was just wondering if there’s an expected release date? Thank you!
From Geraldine_VdAuwera on 2018-03-08
Hi Katie, we don’t have a hard date scheduled but I can tell you we’re in the final stages of picking names for the different tools involved in the workflow, so it shouldn’t be long before we’re able to release a first official version into the world.
From Katie on 2018-03-08
Thank you!
From Oprah on 2018-03-29
Any updates? It’s been 3 weeks …
From Geraldine_VdAuwera on 2018-03-31
Oh yes sorry, it has been released publicly (still BETA though), in [GATK 4.0.3.0](https://github.com/broadinstitute/gatk/releases/tag/4.0.3.0). Docs aren’t up yet but the software is there for you to play with!
From shrutiisarda on 2018-05-09
Hello,
Is there a timeline established for the CNN based filtering approach to replace VQSR in the published best practices workflows yet? Just curious to know when the fully implemented versions roll out. :)
From Geraldine_VdAuwera on 2018-05-09
@shrutiisarda Not yet, there’s still a lot of testing to be done on scalability and robustness of the tools. We want to put a lot more data through this process before we declare it suitable as a replacement for VQSR. That being said people who can’t do VQSR anyway probably have a lot to gain from trying it out. We plan to produce some documentation in the near future to describe how to run these tools.
From shrutiisarda on 2018-05-23
@Geraldine_VdAuwera
Thanks for the info!
From Chris_M on 2018-07-25
Can you please give us an update on the status and maybe a release date? I have several full genomes in the pipeline and wondering if I should wait for the release.
From shlee on 2018-07-25
Hi @Chris_M,
As with any new analysis, anticipating future production status is difficult. The current recommended filtering Best Practice is still VQSR. If you need to filter your callset now, then please use VQSR. We would love for you to also try out the CNN workflow and to share with us what you think. If you try out the workflow now, you’ll be familiar with the steps and considerations so that later, when CNN gets the production-stamp-of-approval, you’ll be able to run the analysis smoothly.
From lailai on 2018-08-31
Thank you!
Report abuse